Overzicht hartafwijkingen
Ongeveer 1 op de 100 kinderen wordt geboren met een hartafwijking. De ernst van de afwijking en het behandelplan verschilt per type afwijking.

Ongeveer 1 op de 100 kinderen wordt geboren met een hartafwijking. De ernst van de afwijking en het behandelplan verschilt per type afwijking.
Bij aortaklepstenose is de klep tussen de linkerkamer en lichaamsslagader (aorta) verdikt of vergroeid. Vaak bestaat ze slechts uit 2 in plaats van 3 klepblaadjes (bicuspide klep). De klep opent onvoldoende om het bloed naar het lichaam te doen stromen. Hierdoor moet de linkerkamer meer druk uitoefenen om het bloed toch door die vernauwde klepopening te stuwen. De continu verhoogde druk veroorzaakt een verdikking van de hartspier (vergelijkbaar met atleten die voortdurend grote inspanningen leveren). Uiteindelijk geeft dit op lange termijn vermoeidheid en aftakeling van de hartspier.
Bij een milde vernauwing hebben kinderen met deze afwijking nauwelijks klachten. Er is bijna altijd een hartgeruis dat uiteindelijk tot verder onderzoek zal leiden. Bij een ernstige vernauwing is er wel kortademigheid. Er zijn vaak klachten van hartkloppingen, pijn op de borstkas bij inspanning en flauwvallen.
De diagnose wordt verkregen door echocardiografie. Deze brengt de verdikte klep in beeld. Doppler-technieken laten ons toe het drukverschil over de klep te meten. Bij de verdere opvolging wordt regelmatig een inspanningstest uitgevoerd om problemen met de hartdoorbloeding op te sporen.
Een behandeling wordt ingesteld vanaf een drukverschil van 50 mm Hg of indien er klachten zijn die toegeschreven kunnen worden aan de klepverdikking. Vaak kan een behandeling bestaan uit hartkatheterisatie met ballondilatatie. Deze wordt uitgevoerd onder algehele anesthesie. Het risico van de ingreep is relatief laag en er zijn weinig complicaties. Alleen bij pasgeborenen is de ingreep iets delicater. De resultaten zijn behoorlijk doch er kan opnieuw stenose ontstaan die een tweede of derde dilatatie nodig maken. Bij onvoldoende resultaat of bij het ontstaan van belangrijke insufficiëntie (lek) is chirurgisch ingrijpen noodzakelijk. Er zijn verschillende technieken mogelijk, afhankelijk van de leeftijd van de patiënt en de ernst van de aandoening. Soms volstaat het om de klep chirurgisch te openen (commisurotomie) maar in vele gevallen is vervanging noodzakelijk. Meestal wordt hiervoor een Ross-operatie uitgevoerd, waarbij de eigen pulmonalisklep als aortaklep wordt gebruikt.
Bij aortaklepstenose is voorzichtigheid geboden bij het uitoefenen van sport. Competitiesport is uitgesloten.
Tijdens de zwangerschap kunnen er problemen optreden. Het is dus belangrijk de aortastenose te behandelen vooraleer aan een zwangerschap te beginnen. Indien er een mechanische klep is ingeplant, is antistollingstherapie noodzakelijk. Deze kan echter belangrijke aangeboren afwijkingen geven bij de foetus en wordt daarom voor de conceptie gestopt. Bij het overschakelen naar een andere, minder toxische therapie is het nodig de gevaren en de voordelen nauwkeurig tegen elkaar af te wegen.
Hartekind Pepijn heeft Aortaklepstenose, lees hier zijn verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
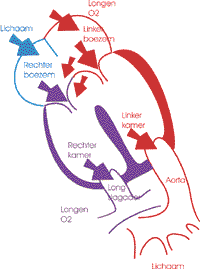
Bij het AVSD bestaat er een grote opening in het midden van het hart. Deze opening kan zich bevinden tussen kamers en boezems (klassieke AVSD), enkel tussen de kamers (AVSD type VSD), of enkel tussen de boezems (ASD type 1). Er is ook steeds een afwijking aan de kleppen tussen de boezems en kamers (vooral de mitralisklep). Deze liggen immers midden in de opening.
Het bloed stroomt doorheen de openingen van het linkerhart naar het rechterhart. Dit resulteert in een overdreven bloedstroom naar de longen. Uitgezonderd bij het ASD type 1 zijn er reeds klachten op jonge leeftijd (enkele weken tot maanden na de geboorte): snelle ademhaling en zweten bij drinken, gevolgd door slechter drinken, slechte gewichtstoename en bleke of grauwe kleur. Meestal is er een hartgeruis. Indien er niet wordt ingegrepen, zullen de klachten alleen maar toenemen.
Spontane sluiting van deze defecten is zeer uitzonderlijk en meestal is chirurgische sluiting nodig. Indien deze niet tijdig gebeurt, bestaat er een risico op overlijden of op beschadiging van de longvaten (pulmonalehypertensie) waardoor een operatie uiteindelijk onmogelijk wordt.
De chirurgische ingreep gebeurt bij voorkeur voor de leeftijd van 6 maanden. De openingen tussen boezems en kamers worden gesloten en de afwijkende kleppen hersteld.
Kinderen met Down syndroom hebben meer dan 25% kans op een AVSD. Omdat hun longvaten veel gevoeliger zijn voor de verhoogde longdruk wordt de leeftijd voor operatie verlaagd naar 3 à 4 maanden. Bij kinderen met Down ontbreekt het hartgeruis dikwijls.
Kinderen met een ASD type 1 hebben veel minder klachten en veel minder kans op pulmonale hypertensie en longvaatbeschadiging. Een chirurgische correctie tussen 2 en 5 jaar lijkt hier ideaal.
Het operatief risico is beperkt, zeker indien de ingreep tijdig gebeurt. Restletsels zijn evenwel niet ongewoon. Het risico op een pacemaker na de operatie is iets hoger dan bij sluiting van een gewoon VSD. In een beperkt aantal gevallen blijft de mitralisklep lekken en moet een tweede keer hersteld worden. In sommige gevallen zal later zelfs een kunstklep moeten ingeplant worden.
Hartekind Levi heeft o.a. een incompleet AVSD, lees hier zijn verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
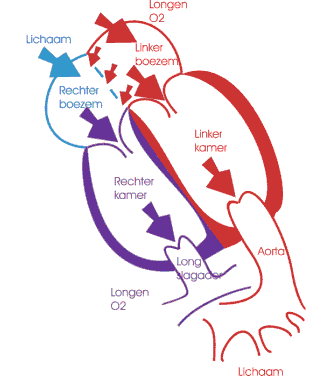
Een atriaal septum defect is een opening in het tussenschot tussen de linker- en de rechtervoorkamer. Er wordt een onderscheid gemaakt tussen 3 types ASD.
Het meest voorkomende type is het ASD II of ASD secundum. Deze verbinding is een restant van het leven in de baarmoeder. Normaal zal deze opening, ook foramen ovale genoemd, na de geboorte worden afgesloten door een vlies dat als een deksel de opening tussen de voorkamers afsluit. Gebeurt dit niet, dan blijft de verbinding bestaan. Indien een ASD II tijdens de eerste levensjaren wordt opgemerkt, bestaat er een kans dat het toch nog spontaan zal sluiten.
Een ASD I of ASD primum ontstaat wanneer het tussenschot van de voorkamers abnormaal ontwikkelt. De opening tussen de voorkamers ligt dicht tegen de mitralisklep aan en dit defect gaat bijna steeds gepaard met een lek ter hoogte van deze klep (mitralisinsufficiëntie). Een spontane sluiting van een ASD I valt niet te verwachten.
Ook het sinus venosus ASD ontstaat door een abnormale aanleg van de scheidingswand tussen de voorkamers. Deze opening is echter veel hoger gelegen, tegen de uitmonding van de grote lichaamsaders aan. De rechter longvenen monden steeds abnormaal uit. Een spontane sluiten van een sinus venosus ASD valt niet te verwachten.
Indien er een opening bestaat tussen de beide voorkamers, kan het bloed rechtstreeks van de linker- naar de rechtervoorkamer stromen. Hoe groter het defect is, hoe groter de shunt. Door deze supplementaire hoeveelheid bloed zal de rechter kamer beginnen uitzetten en zullen ook de longen overvuld geraken. Kinderen met een ASD hebben op deze manier vaker longinfecties dan anderen en ook de gewichtsevolutie verloopt minder vlot. Op babyleeftijd kan een flinke overbelasting van de rechterkamer aanleiding geven tot moeilijk drinken, een snelle ademhaling en zweten. Het optreden van deze klachten zal meestal de aanleiding zijn om het ASD te behandelen, maar meestal kan gewacht worden tot de leeftijd van 3 à 5 jaar. Indien de rechterkamer gedurende lange tijd overbelast is geweest, kan ze verdikken en minder goed functioneren. Een langdurige overvulling van de longcirculatie kan leiden tot een verhoogde bloeddruk in de longen.
Een klein ASD II geeft weinig klachten en wordt vaak toevallig ontdekt. Een behandeling is zelden nodig. Op volwassen leeftijd zijn er echter uitzonderingen. Het ASD vormt immers een verbinding tussen het linker- en het rechterhart. Zo kunnen niet alleen bloed maar ook embolen in de systeemcirculatie geraken. Om deze reden wordt een klein ASD II toch gesloten bij diepzeeduikers en bij ritmestoornissen.
Indien bij klinisch onderzoek een ASD vermoed wordt, kan de diagnose worden gesteld door middel van echocardiografie. Hiermee kan het type ASD worden bepaald en kan een evaluatie gebeuren van de overbelasting van de rechterkamer.
Er zijn 2 mogelijke behandelingen voor ASD: chirurgische sluiting of het plaatsen van een paraplu door middel van hartkatheterisatie. Welke techniek wordt gebruikt hangt af van het type ASD, de grootte en de ligging. Sluiten via hartkatheterisatie is mogelijk vanaf een leeftijd van ongeveer 3 jaar of soms jonger indien te klachten te ernstig zijn. Om een goede aanhechting van de paraplu mogelijk te maken is het nodig dat rondom het ASD een voldoende grote rand weefsel ligt. Zowel bij ASD I als bij sinus venosus ASD is dit niet het geval.
Hartekind Jurre heeft een ASD, lees hier zijn verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
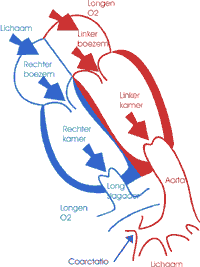
Een coarctatio aortae is een vernauwing op de grote lichaamsslagader ter hoogte van de ductus arteriosus, juist onder de aftakking van de bloedvaten naar de linker arm. Vaak gaat deze aandoening gepaard met een smallere (hypoplastische) aortaboog. Door de vernauwing op deze belangrijke slagader komt de bloedvoorziening van onderste lichaamshelft in het gedrang. Bovendien wordt de weerstand waartegen de linkerkamer moet pompen groter zodat de voorkamer gaat verdikken (hypertrofie) en ontstaat er een hoge bloeddruk.
Coarctatio aortae kan zich onder verschillende vormen en op verschillende leeftijden presenteren. Op jonge zuigelingenleeftijd kan een coarctatio aortae voor grote problemen zorgen op het ogenblik dat de ductus sluit: de vernauwing in de aorta wordt nog meer uitgesproken en er valt een alternatieve bevloeiingsweg naar de onderste lichaamshelft weg. De baby begint sneller te ademen en slechter te drinken. Als de toestand niet tijdig wordt onderkend kan dit leiden tot een acute decompensatie die snel behandeld moet worden. Coarctatio aortae kan ook op latere leeftijd worden ontdekt. De vernauwing is dan minder uitgesproken en het lichaam heeft kleine zijvertakkingen (collateralen) gevormd die de onderste lichaamshelft bevloeien.
De diagnose wordt meestal gesteld naar aanleiding van een vergrote hartschaduw op een RX van de longen, slechte polsslag in de liezen en hoge bloeddruk. Indien de diagnose van coarctatio aortae klinisch wordt vermoed, zal in de eerste plaats een bloeddrukmeting worden uitgevoerd. Hierbij zal de bloeddruk aan de armen duidelijk hoger zijn dan aan de benen. De diagnose kan worden bevestigd door middel van echocardiografie. Hiermee kan men niet enkel de plaats en de lengte van de vernauwing in het licht stellen maar kan men ook de verdikking van de hartspier evalueren. Bovendien kunnen geassocieerde hartafwijkingen worden uitgesloten. Vooral bij grotere kinderen, volwassen en in het geval van recoarctatio is het niet steeds mogelijk de coarctatio perfect te visualiseren. Een gespecialiseerde magnetische resonantie scan (MRI) kan dan bijkomende informatie opleveren.
Indien bij een jonge zeer zieke baby de diagnose van coarctatio aortae wordt gesteld, zal in de eerste plaats getracht worden de ductus arteriosus opnieuw te openen door middel van medicatie. Eens het kind stabiel is, zal steeds worden overgegaan tot het chirurgisch wegnemen van de vernauwing (coarctectomie). Na de ingreep kan de hoge bloeddruk gedurende enkele maanden blijven bestaan zodat medicatie nog enige tijd noodzakelijk blijft.
Ook bij oudere kinderen wordt, gezien de relatieve eenvoud en de goede resultaten van de ingreep, meestal een chirurgisch wegname uitgevoerd. Er kan echter in bepaalde gevallen ook worden overgegaan tot een hartkatheterisatie met ballondilatatie van de vernauwde aorta.
Bij volwassen patiënten worden zowel de chirurgische coarctectomie als de hartkatheterisatie uitgevoerd.
Het is belangrijk dat patiënten na de coarctectomie medisch gevolgd worden. Er kan immers steeds een recoarctatio optreden, die op zijn beurt moet worden behandeld. Meestal wordt dan een hartkatheterisatie uitgevoerd waarbij de vernauwing wordt opgerokken dmv een ballondilatatie. Eventueel wordt een stent geplaatst.
Hartekind Silke is geboren met coarctatio aortae, lees hier haar verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Men spreekt van double outlet rechterventrikel wanneer het grootste deel van de oorsprong van de grote bloedvaten (aorta en longslagader) ontspringt boven de rechterkamer. Deze afwijking gaat steeds gepaard met andere defecten in het hart. Naast de meer zeldzame complexe hartafwijkingen (die hier niet afzonderlijk worden besproken), worden 3 grote groepen beschreven. De klinische tekens en behandeling zijn sterk verschillend naargelang het type.
Initieel zijn er meestal weinig klachten. Als de longdrukken na de geboorte systematisch beginnen te dalen, zal er relatief meer bloed stromen naar de longen. Dit kan leiden tot snel ademen (tachypneu), slecht drinken en een onbevredigende gewichtsevolutie. Bij klinisch onderzoek is er steeds een geruis hoorbaar. De behandeling bestaat uit chirurgie waarbij het VSD wordt gesloten door middel van een patch Hierbij wordt een soort tunnel van de linkerkamer naar de aorta gecreëerd. De resultaten na deze ingreep zijn goed. Wel moet men steeds bedacht zijn op het optreden van een subaortale vernauwing. Nauwkeurige opvolging is dus noodzakelijk.
Net zoals bij Tetralogie van Fallot is de ernst en het tijdstip van de klachten afhankelijk van de ernst van de pulmonalisstenose. De klachten zijn afhankelijk van de graad en plaats van de vernauwing naar de longen toe. Hoe nauwer de uitgang naar de longen, hoe vroeger en hoe ernstiger blauwheid zal optreden. Het tijdstip van de behandeling hangt af van de ernst van de blauwheid. Onder de leeftijd van 3 maanden wordt geopteerd voor een tijdelijke oplossing zoals de Blalock-Taussig shunt (chirurgisch aanleggen van verbindingsvat tussen lichaamsslagader en longslagader). Boven de leeftijd van 3 maanden wordt meestal gekozen voor een totale correctie. Hierbij wordt het VSD gesloten door middel van een patch. De longslagader wordt met een klepdragende buisprothese verbonden met de rechterkamer (Rastelli procedure). Het risico van de ingreep is matig maar restletsels zijn niet zeldzaam. Het kan gaan om een vernauwing onder de aorta (subaortale stenose), om een vernauwing van de pulmonaalklep of vernauwingen van de longslagaders. Op oudere leeftijd moet de buisprothese vaak worden vervangen.
Dit type uit zich meestal enkele dagen tot weken na de geboorte door klachten veroorzaakt door de coarctatio, een vernauwing op de grote lichaamsslagader. De baby begint sneller te ademen en slechter te drinken. Als de toestand niet tijdig wordt onderkend kan dit leiden tot een acute decompensatie die snel behandeld moet worden.
Bij ernstig hartfalen kan in eerste instantie getracht worden de baby te stabiliseren door het openhouden van de ductus arteriosus door middel van prostaglandines. Nadien kan worden overgegaan tot een totaal herstel. Dit is een zeer complexe en risicovolle chirurgische ingreep waarbij verschillende technieken worden gecombineerd: 1) VSD sluiting door middel van een patch; 2) arteriële switch proceduremet omwisselen van de grote vaten de reïmplantatie van de kransslagaders op de aorta; 3) coarctectomie. De risico’s bij deze ingreep zijn relatief hoog en het postoperatief verloop is vaak langdurig en moeilijk. Meestal hebben de kinderen nadien nood aan tijdelijke medicamenteuze ondersteuning. Toch is de levenskwaliteit en de levensverwachting later goed. Mogelijke complicaties later bestaan uit het optreden van ritmestoornissen, subaortale obstructie of recoarctatio waarvoor reïnterventie soms noodzakelijk is.
Hartekind Kayleigh heeft een DORV, lees hier haar verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Dubbele Discordantie (ook wel Congenitaal gecorrigeerde transpositie van de grote vaten) is een zeldzame aangeboren hartafwijking waarbij er een verkeerde verbinding bestaat op 2 opeenvolgende niveaus in het hart:
Het gevolg is dat het zuurstofarme (“blauwe”) bloed zoals het hoort naar de longen stroomt en het zuurstofrijke (“rode”) bloed naar het lichaam. Het probleem is echter dat de rechterkamer nu naar de systeemcirculatie (met hogere bloeddruk) moet pompen. Structureel is die rechterkamer daar niet voor gemaakt. Dit kan op termijn leiden tot “slijtage” en hartfalen. Dubbele discordantie kan gepaard gaan met andere defecten in het hart of deel uitmaken van een complex hartgebrek.
De behandeling van congenitaal gecorrigeerde transpositie van de grote vaten is afhankelijk van het tijdstip van diagnose en de hartafwijkingen die een kind heeft.
De prognose van congenitaal gecorrigeerde transpositie van de grote vaten is dus sterk verschillend en afhankelijk van zowel hartfunctie, geassocieerde afwijkingen en aard van de behandeling. Soms is het nodig over te gaan tot harttransplantatie.
'Geef Hartekind een gezicht' en deel je verhaal.
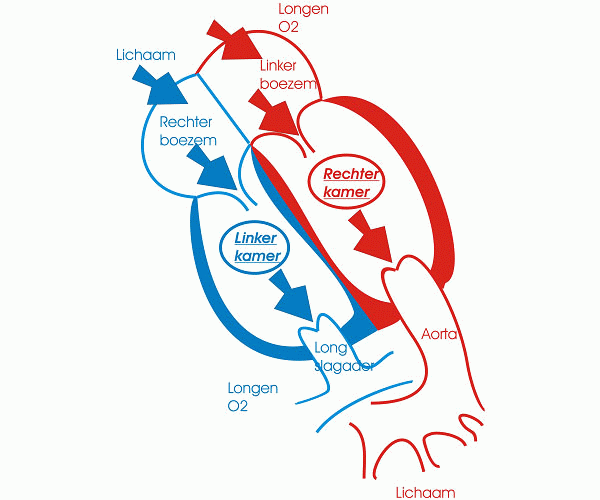
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
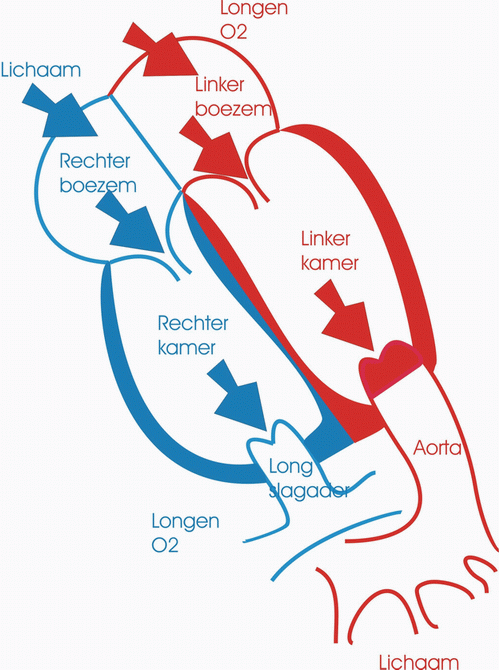
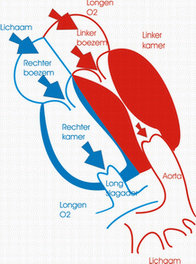
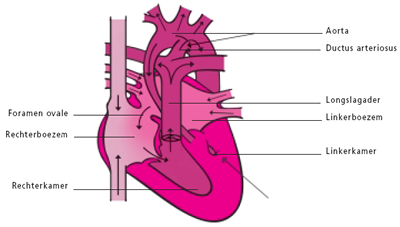
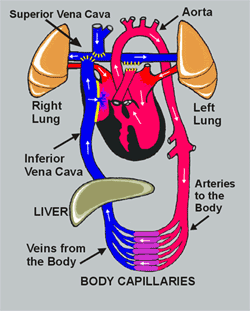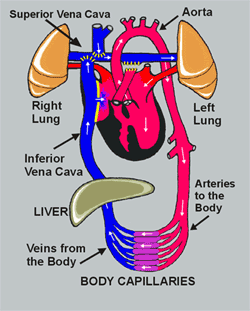
De ductus arteriosus of ductus van Botall is een bloedvat dat de longslagader (a. pulmonalis) verbindt met de lichaamsslagader (aorta). Hij speelt vooral een belangrijke rol tijdens de zwangerschap.
Tijdens het normale leven zorgen de longen voor de opname van zuurstof en de afvoer van koolstofdioxine. Het zuurstofarme bloed dat vanuit het lichaam in de rechtervoorkamer aankomt, vertrekt vanuit de rechterkamer via de longslagader naar de longen waar de zuurstofuitwisseling plaatsvindt. Het zuurstofrijke bloed zal daarna via de linkervoorkamer en linkerkamer naar het lichaam worden gevoerd. Tijdens het leven in de baarmoeder echter wordt deze rol van zuurstofuitwisseling overgenomen door de moederkoek. De bloeddruk in de longen is op dat ogenblik zeer hoog en volgens de wetten van de fysica neemt het bloed de weg van de minste weerstand: het zuurstofarme bloed stroomt van het rechterhart niet naar de longen maar via het foramen ovale en de ductus arteriosus naar de aorta en zo naar de moederkoek. Onmiddellijk na de geboorte nemen de longen hun taak van gasuitwisselaar op. De bloeddruk in de longcirculatie zakt drastisch en het zuurstofarme bloed zal nu wel via de longslagader naar de longen stromen. Op dat ogenblik is de taak van de ductus arteriosus als “binnenweg” voor het bloed uitgespeeld. Normalerwijze zal dit bloedvat dan ook onder invloed van verscheidene fysische en chemische processen spontaan sluiten.
Meestal sluit de ductus arteriosus binnen de eerste 3 levensdagen. In enkele specifieke gevallen echter gebeurt dit niet. Zo komt een persisterende ductus arteriosus (open openblijven van de ductus arteriosus) bijvoorbeeld vaak voor bij prematuur geboren kinderen. De mechanismen die zorgen voor het sluiten van de ductus zijn bij hen immers nog onvoldoende ontwikkeld. Er blijft dus een verbinding bestaan tussen de lichaams- en de longslagader.
Na de geboorte daalt de bloeddruk in de longen zodat deze lager wordt dan de bloeddruk in de lichaamscirculatie. Het zuurstofarme bloed dat vertrekt vanuit de rechterkamer zal dus probleemloos naar de longslagader en naar de longen stromen. Het zuurstofrijke bloed dat vanuit de linkerkamer in de aorta stroomt zal echter door de lagere bloeddruk in de longen (opnieuw via het principe van de weg van de minste weerstand), voor een deel via de ductus arteriosus opnieuw naar de longen gaan. De longen raken op deze manier te veel gevuld met bloed. Dat geeft aanleiding tot zuurstofnood. Als de problemen te groot zijn moeten deze pasgeboren baby’s behandeld worden. Is er geen zuurstofnood, dan kan worden afgewacht. De ductus arteriosus kan immers nog spontaan sluiten tot 6 maanden na de geboorte.
Persisterende ductus arteriosus kan ook worden vastgesteld bij oudere kinderen. Soms gaat het om patiënten die na de geboorte medische problemen hebben gehad of om patiënten die een aangeboren hartafwijking hebben. Vaak echter kan er geen duidelijke oorzaak van het openblijven van de ductus worden achterhaald. De diagnose van persisterende ductus arteriosus wordt bij oudere kinderen bijna steeds gesteld naar aanleiding van een toevallig ontdekt hartgeruis. Meestal hebben de zij geen klachten, maar patiënten bij wie een grote hoeveelheid bloed (= grote shunt) naar de longen terugstroomt, hebben vaak last van luchtweginfecties . Zij komen meestal ook slecht aan in gewicht.
Als de diagnose van persisterende ductus arteriosus wordt vermoed, kan er het beste een echocardiogram worden gemaakt. Hierbij kan dit bloedvat niet enkel worden gevisualiseerd, maar kan er ook worden geëvalueerd hoeveel bloed opnieuw naar de longen stroomt. Bovendien kunnen eventuele complicaties worden ontdekt.
Als een grote ductus arteriosus niet spontaan sluit, is het belangrijk dat deze kunstmatig gesloten wordt. Via dit bloedvat immers stromen grote hoeveelheden bloed terug naar de longen. Deze raken “overstroomd” wat aanleiding geeft tot zeer vaak voorkomende longinfecties. Als de situatie lang aanhoudt, kan dit bovendien aanleiding geven tot pulmonale hypertensie (gefixeerde hoge bloeddruk in de longvaten). In zeldzame gevallen kan de persisterende ductus arteriosus ook aanleiding geven tot hartinfectie bij bloedinfectie of tot trombose.
Als een pasgeboren baby zuurstofnood heeft omwille het openblijven van de persisterende ductus arteriosus kan getracht worden de ductus te sluiten met medicijnen. In het Universitaire Ziekenhuis van Gent wordt vooral gebruik gemaakt van indomethacine (Indocid), maar andere centra maken vaak ook gebruik van ibuprofen. Het succes van deze behandeling is wisselend. Kleine prematuren en erg zieke kinderen reageren duidelijk minder goed.
Als medicatie niet aanslaat en de baby veel last heeft, moet soms worden overgegaan tot een chirurgische sluiting (ligatie).
Bij oudere kinderen is het niet meer mogelijk de ductus via medicijnen te sluiten. Een hartoperatie is bij deze patiënten echter zelden noodzakelijk. Sinds een tiental jaren kan de ductus arteriosus worden gesloten doormiddel van hartkatheterisatie. Deze techniek is mogelijk vanaf een gewicht van 5 kg.
Als de ductus arteriosus nog open is, dan moet bij het vullen van tanden of het verwijderen van tandsteen éénmalig een dosis antibioticasiroop worden ingenomen. Doet dit een half uur voor de ingreep. Breng uw tandarts op de hoogte zodat dit in het dossier van uw kind kan worden genoteerd. Ook bij heelkundige ingrepen waarbij er een risico bestaat dat kiemen in de bloedbaan komen, moet voor de ingreep antibiotica worden genomen.
'Geef Hartekind een gezicht' en deel je verhaal.
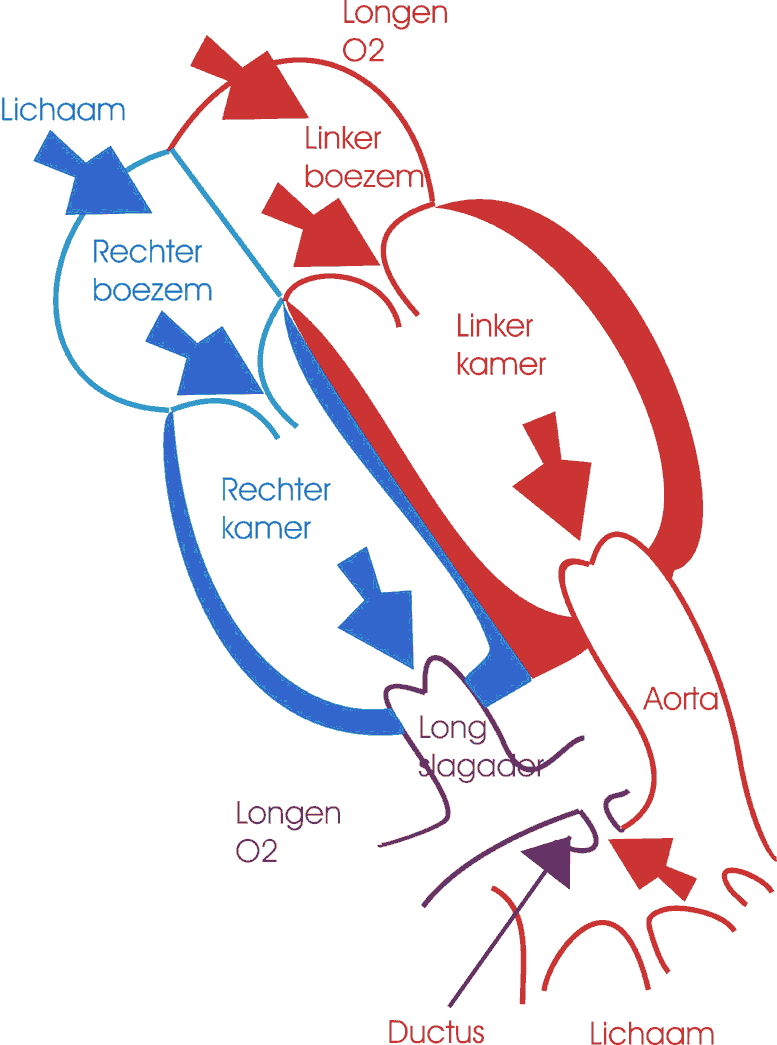
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Endocarditis is de latijnse naam voor endocard-ontsteking. Het endocard is het binnenste vliesje van de hartspier. Het is de cellaag die de bloedstroom begrenst. Deze ontsteking kan zowel veroorzaakt worden door een infectie (meest voorkomend) als door iets anders (bijvoorbeeld een auto-immuun reactie).
Infectieuze endocarditis is een ontsteking van het endocard door een “besmettelijk” organisme. Dit kan een schimmel, een amoebe, een virus of een bacterie zijn. Vooral de bacteriële endocarditis is het meest te vrezen. Endocarditis kan optreden als een verwikkeling na heelkunde of veroorzaakt worden door bacteriën (“microben”) die zich vanuit het lichaam via de bloedbaan verspreiden. De besmetting zal bij voorkeur ontstaan op plaatsen van littekenweefsel dat ontstaat door hevige wervelingen van het bloed (ter hoogte van een VSD, ter hoogte van een zieke klep, of in een open ductus arteriosus).
Er is alleen een grote kans op besmetting als er veel bacteriën in de bloedbaan aanwezig is. Dit kan gebeuren bij de aanwezigheid van belangrijke etter-haarden (abces), en vooral op het ogenblik van het insnijden of manipuleren. Voorbeelden zijn tandontstekingen, ontstoken poliepen en amandelen of bijvoorbeeld blindedarmontsteking. Deze ontstekingen worden meestal door een bepaalde kiem veroorzaakt, zodat aan de hand van de lokalisatie bepaald kan worden welk antibioticum het meest geschikt is.
Bij personen met een hartafwijking en zeker als er bij een operatie vreemd materiaal (klep, conduit, …) werd geplaatst, zal men trachten de kans op endocarditis te verlagen door het geven van een hoge dosis van een aangepast antibioticum. Men spreekt dan van endocarditis “profylaxe” of Anti-Osler profylaxis (het vooraf behandelen vooraleer er een besmetting is). Profylaxe wordt enkel gegeven op het ogenblik van een medische ingreep waarbij de kans op het mobiliseren van grote hoeveelheden bacteriën bestaat, zoals bij het trekken van een besmette kies, het trekken van adenoïde woekeringen (poliepen of amandelen), het uitkrabben van etter-nestjes in het tandvlees, enzovoorts.
Endocarditis profylaxe bestaat dan ook uit het heel kort (vaak slechts één enkele hoge dosis) geven van een aangepast antibioticum, meestal Ampicilline. Bij buikbesmettingen wordt Gentamycine toegevoegd. De belangrijkste voorzorgsmaatregel is en blijft echter het vermijden van ettercollecties ergens in het lichaam. Men noemt dit het vermijden en tijdig onderkennen van “focale” infecties. Voorbeelden hiervan zijn tandcaries, chronische sinusitis of ontsteking van het tandvlees. Een goede tand en mondhygiëne is dus een must voor iedereen met een aangeboren hartafwijking. Zesmaandelijkse controle bij de tandarts wordt hier best strikt opgevolgd, naast een dagelijkse poetsbeurt. Bij voorkeur poetst men zijn tanden na iedere maaltijd. Zowel piercing als tatoeage zijn een belangrijke bron van infecties. Ze zijn dan ook sterk af te raden.
'Geef Hartekind een gezicht' en deel je verhaal.
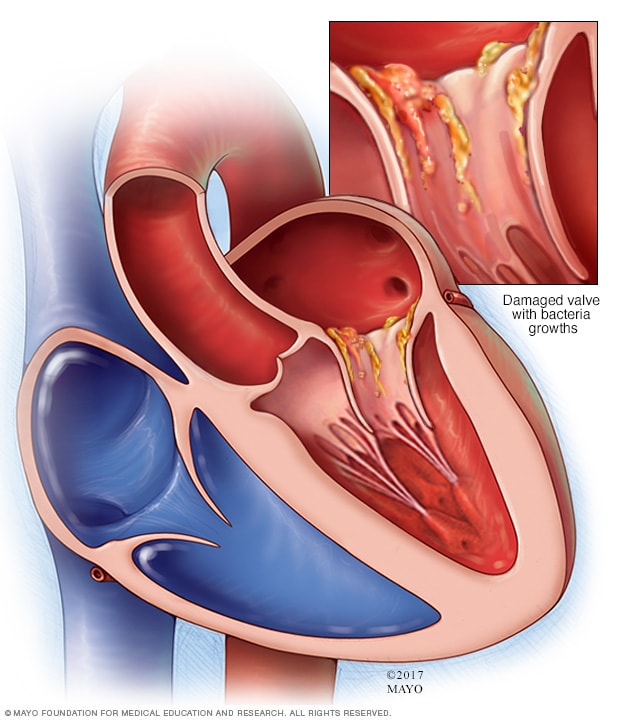
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Gedilateerde cardiomyopathie is een afwijking van de hartspier, waarbij één of beide kamers uitgezet zijn en een verminderde functie vertonen. Bij ernstige aantasting zijn de kinderen kortademig en snel moe bij inspanning. Bij verdere evolutie treden er vaak ritmestoornissen op.
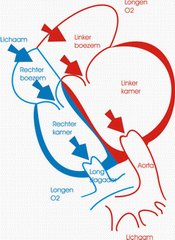
De oorzaak van gedilateerde cardiomyopathie kan zeer verscheiden zijn en zowel verworven als aangeboren. Bij pasgeborenen en zuigelingen wordt de aandoening meestal veroorzaakt door een infectie (viraal, bacterieel, …) en men spreekt dan van myocarditis. Deze vorm van cardiomyopathie kan, eens de acute fase voorbij, recupereren. Chronisch gebruik van cardiotoxische medicatie kan eveneens gedilateerde cardiomyopathie geven. Dit is het beste gekend bij bepaalde soorten kankertherapie. Kinderen met deze vorm van behandeling worden dan ook regelmatig echocardiografisch gevolgd om aantasting tijdig op te sporen en een adequate behandeling in te stellen. Ook indien er langdurig snelle hartritmes blijven bestaan (bijvoorbeeld chronische supraventriculaire tachycardie) kan de linkerhartkamer dilateren. Naast deze verworven vormen van gedilateerde cardiomyopathie bestaan er ook aangeboren vormen. Vaak komt deze afwijking voor bij verschillende leden van één familie. Bij een eerste groep patiënten gaat het om een geïsoleerde hartafwijking zonder andere problematiek. Er zijn op dit ogenblik verschillende gendefecten gekend die deze aandoening kunnen veroorzaken. Ook bepaalde stofwisselingsziekten (metabole afwijkingen) kunnen gepaard gaan met gedilateerde cardiomyopathie. Deze kinderen hebben meestal problemen op niveau van verschillende orgaansystemen.
De diagnose van gedilateerde cardiomyopathie wordt gesteld door middel van echocardiografie. Om de juiste oorzaak van de problematiek op te sporen worden eveneens verscheidene uitwerkende onderzoeken uitgevoerd. Deze bestaan meestal uit een uitgebreide bloedname, urinecollectie, neurologisch en genetisch onderzoek, en eventueel MRI. In bepaalde gevallen zal er worden overgegaan tot hartkatheterisatie met biopsie (klein stukje hartspierweefsel wordt weggenomen).
Lees het verhaal van Niek, geboren met gedilateerde cardiomyopathie.
Gelukkig zijn ritmestoornissen bij het kind veel zeldzamer dan bij volwassenen. Bij kinderen zonder onderliggende hartafwijking en zonder beladen familiale geschiedenis zijn ritmestoornissen meestal ongevaarlijk en is de kans op genezing vrij groot. Bij kinderen met ritmestoornissen bij dichte familieleden of onderliggende hartafwijking zijn de risico’s veel groter en is genezing veel moeilijker.
De meest frequente ritmestoornissen bij kinderen zijn eenvoudig in te delen.
Methodes om tot een correcte diagnose te komen:
Electrofysiologisch onderzoek: hartcatheterisatie waarbij specifiek ritmestoornissen worden opgewekt om hun aard en lokalisatie beter te begrijpen.
'Geef Hartekind een gezicht' en deel je verhaal
Hypertrofe cardiomyopathie is een afwijking van de hartspier, waarbij de wand van één of beide kamers verdikt is en soms een verminderde functie vertoont.
Meestal vertonen kinderen geen symptomen. Bij ernstige aantasting echter zijn ze moe, kortademig en hebben een verminderd inspanningsvermogen. Bij verdere evolutie treden er vaak ernstige ritmestoornissen op.
Hypertrofe cardiomyopathie is meestal aangeboren. Vaak komen er verscheidene patiënten voor in één familie. Er zijn op dit ogenblik verschillende gendefecten gekend die deze aandoening kunnen veroorzaken. Patiënten kunnen zowel geïsoleerd hartlijden hebben als associaties met andere afwijkingen. Bekende genetische afwijkingen met o.a. hypertrofe cardiomyopathie zijn de Friedraich’s ataxie en het Noonan syndroom. Ook bepaalde stofwisselingsziekten (metabole afwijkingen) kunnen gepaard gaan met hypertrofe cardiomyopathie. Deze kinderen hebben meestal problemen op niveau van verschillende orgaansystemen. Eén van de meer goedaardige vormen van hypertrofe cardiomyopathie komt voor bij pasgeborenen van moeders met diabetes mellitus.
De diagnose van hypertrofische cardiomyopathie wordt gesteld door middel van echocardiografie. Om de juiste oorzaak van de problematiek op te sporen worden eveneens verscheidene uitwerkende onderzoeken uitgevoerd. Deze bestaan meestal uit een uitgebreide bloedname, urinecollectie, neurologisch en genetisch onderzoek, en eventueel MRI. In bepaalde gevallen zal er worden overgegaan tot hartkatheterisatie met biopsie (klein stukje hartspierweefsel wordt weggenomen).
De prognose van hypertrofische cardiomyopathie is afhankelijk van de oorzaak doch meestal is het proces onomkeerbaar. De behandeling is voornamelijk medicamenteus. Is de evolutie niet te stoppen en nemen de klachten toe, wordt overgegaan tot harttransplantatie.
'Geef Hartekind een gezicht' en deel je verhaal
Het hypoplastisch linkerhart syndroom is een zeer ernstige aangeboren hartafwijking waarbij de linker helft van het hart (de helft die zorgt voor de lichaamscirculatie) veel te weinig ontwikkeld (hypoplastisch) is. De mitralisklep en de aortaklep openen nauwelijks of niet en de wand van de linkerkamer is sterk verdikt en stijf. Dit resulteert in een klein, niet-functionerend linkerkamertje. Het bloed kan enkel naar het lichaam stromen vanuit de rechterkamer en zo via de longslagader en de ductus arteriosus naar de aorta. Dit leidt tot zeer ernstige problemen kort na de geboorte, het ogenblik waarop de ductus arteriosus sluit. De pasgeborene wordt vaak acuut slecht met een bleek-grauwe kleur en een snelle ademhaling. Indien er niet snel wordt ingegrepen gaat de baby zienderogen achteruit.
De eerste stap in de behandeling bestaat erin de ductus arteriosus weer te open door middel van intraveneus toegediende medicatie. Op deze manier kan het bloed weer, via een omweg, naar het lichaam stromen. In de meeste gevallen is bijkomende medicatie nodig om de hartspier te versterken, om de verzuring van het bloed te neutraliseren en om het overtollige lichaamswater uit te plassen. Bovendien moet men de kinderen in een aantal gevallen ondersteunen met kunstmatige beademing.
Na deze stabilisatie fase kan worden overgegaan tot de chirurgische behandeling. Hierbij is het niet mogelijk om het hart opnieuw volledig “normaal te maken”. Via drie verschillende operaties kan men wel de lichaamscirculatie herstellen en het zuurstofrijke en zuurstofarme bloed van elkaar scheiden.
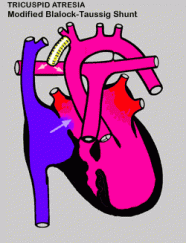
Een eerste operatie wordt kort na de geboorte uitgevoerd, meestal in de eerste levensweek. Deze “Norwood” operatie is en zeer complexe ingreep waarbij de longslagader gebruikt wordt om de uitgang vanuit het hart naar het lichaam te maken. De bloedtoevoer naar de longen gebeurt dan via een kunstmatig aangelegd vat (shunt), ofwel rechtstreeks vanuit de rechterkamer (Sano shunt), ofwel vanuit een zijtak van de aorta (Blalock-Taussig shunt). Deze operatie draagt een zeer groot risico, niet enkel tijdens de ingreep zelf maar gedurende de hele periode tot de volgende chirurgie.
Eenmaal kinderen de eerste operatie doorstaan hebben en wat gegroeid zijn, worden de volgende stappen voorbereid. Het hoofddoel is het bereiken van twee gescheiden circulaties: één circulatie die het zuurstofrijke bloed vanuit de longen naar het lichaam pompt en gebruik maakt van de hartkamer, en de andere die het zuurstofarme bloed vanuit het lichaam naar de longen leidt zonder tussenkomst van de hartkamer. Het bloed van het lichaam wordt door een complexe samenwerking van hart en longen naar de longen “gezogen”. Dat is de reden waarom men er na de geboorte moet voor zorgen dat de druk in de longen laag blijft. Deze lage druk verhindert verdikking van de longvaten zodat deze hun soepelheid behouden. Vooraleer men veilig kan overgaan tot deze chirurgische stappen moeten de druk en de weerstand ter hoogte van de longvaten gemeten worden. Tot nu toe kan dit enkel met hartkatheterisatie. Enkel als de longdruk en longweerstand laag genoeg zijn kan men tot de volgende twee stappenovergaan.
De tweede chirurgische ingreep, de bidirectionele Glenn of Hemi-Fontan procedure, wordt uitgevoerd rond de leeftijd van 3 tot 6 maanden. Er wordt een verbinding gemaakt tussen de bovenste holle ader (vena cava superior) en longslagader. Het zuurstofarme bloed van de bovenkant van het lichaam wordt zo rechtstreeks naar de longen geleid. Het bloed van de onderkant van het lichaam komt nog steeds in de kamer van het hart en wordt gemengd bij het zuurstofrijke bloed vanuit de longen.
De derde en laatste stap, de TCPC of Fontan procedure, wordt uitgevoerd rond de leeftijd van 3 jaar. Er wordt een verbinding aangelegd tussen de onderste holle ader (vena cava inferior) en de longslagader. Al het zuurstofarme bloed wordt nu vanuit het lichaam rechtstreeks naar de longen geleid zonder menging in de kamer. Vanaf nu zijn “linker”- en “rechter” circulatie gescheiden. Initieel laat men nog een kleine verbinding open naar het hart (een fenestratie). Dit is een soort ventiel dat verhindert dat de druk in de aders na de operatie te hoog oploopt. Omdat dit ventielsysteem nog voor een beperkte menging van zuurstofarm en zuurstofrijk bloed zorgt en dus ook een matige cyanose, moet het nadien soms via hartkatheterisatie met een paraplusysteem worden gesloten. In vergelijking met de vorige stappen draagt de TCPC of Fontancirculatie slechts een matig risico.
De uiteindelijke levenskwaiteit van deze kinderen is afhankelijk van een aantal factoren. Initieel worden de overlevingskansen bepaald door de snelheid van diagnose, de graad van decompensatie en recuperatie van de hartfunctie en de aanwezigheid van een niet-restrictief foramen ovale.
Indien alle operaties goed verlopen is, kan men bij vele van deze kinderen een redelijke levenskwaliteit verwachten. Ze zullen wel steeds een of andere vorm van medicatie nodig hebben: aspirine of echte antistolling, initieel ontwateringsmiddelen en hartondersteuning, medicatie tegen ritme-stoornissen. Zij kunnen naar school gaan en inspanningen doen op eigen ritme. Zwangerschap wordt afgeraden wegens risico’s voor moeder en kind, maar is eventueel wel mogelijk indien goed begeleid. Bij sommigen echter zal de hartwerking progressief verminderen. Dit zal zich o.a. vertalen in ritmestoornissen, verminderd inspanningsvermogen of darmklachten. In een aantal van deze gevallen zal enkel harttransplantatie een oplossing kunnen bieden.
Lees het verhaal van Vere en het verhaal van Ann, beide geboren met het hypoplastisch linkerhart syndroom.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Hartekind Mila heeft een hypoplastisch rechter hart syndroom, lees hier haar verhaal
Het Marfan syndroom is een relatief vaak voorkomende erfelijke aandoening die veroorzaakt wordt door een afwijking van het bindweefsel. Het syndroom gaat dan ook niet alleen gepaard met hartafwijkingen maar beïnvloedt bijna het ganse lichaam. Personen met Marfan zijn meestal erg groot en hebben vooral erg lange ledematen. De vingers zijn lang en slank en de gewrichten te beweeglijk. Ook ter hoogte van de rug en de borstkas kunnen vervormingen ontstaan. Bij ongeveer 75% van de patiënten zijn er oogafwijkingen aanwezig, onder de vorm van lensluxaties of bijziendheid.
Cardiale afwijkingen maken meestal ook deel uit van het klinische beeld van Marfan. Bij 75% van de patiënten vindt men afwijkingen ter hoogte van de mitralisklep waarbij de klep prolabeert en lekt. Bij kinderen is dit vaak het eerste teken van cardiale aantasting bij het syndroom van Marfan.
De meest te vrezen complicatie bij het syndroom van Marfan is progressieve uitzetting van de aortawortel. Dit kan aanleiding geven tot aneurysmavorming met mogelijks dissectie (scheur in de wand) of ruptuur (scheur naar buiten toe) tot gevolg. Ook als gevolg van de uitzetting ken er lekkage van de klep ontstaan. De aantasting van de aorta neemt toe met de leeftijd.
De diagnose van Marfan wordt gesteld op basis van vastgelegde klinische criteria. Om aan de diagnose te voldoen is een combinatie van criteria in verschillende orgaansystemen noodzakelijk. Om deze criteria op te sporen zijn een aantal onderzoeken noodzakelijk waaronder een klinisch onderzoek, een echocardiografie, een grondig oogonderzoek, een scanner van de rug en eventueel genetisch onderzoek. Echocardiografie met bepaling van Dopplersnelheden kan de aard en de ernst van het kleplijden aantonen. Dit onderzoek is niet alleen noodzakelijk voor de diagnose, maar ook voor de verdere follow-up van de patiënten.
Aangezien het Marfan syndroom een erfelijke aandoening is, is genetisch advies onontbeerlijk. Het syndroom van Marfan wordt veroorzaakt door mutaties in het fibrilline1 gen (FBN1 mutaties). Er zijn tot op heden al meer dan 500 verschillende FBN1 mutaties ontdekt.
De behandeling van de cardiale afwijkingen van het Marfan syndroom bestaat in eerste instantie uit een medicamenteuze behandeling om de snelheid van uitzetting van de aortawortel tegen te gaan. Indien de diameter een bepaalde grens overschrijdt, is heelkundig ingrijpen noodzakelijk waarbij de aortawortel en – in geval van klepafwijkingen – ook de klep wordt vervangen. De ingreep gebeurt best “electief”, dit wil zeggen op een moment dat rustig van op voorhand kan gepland worden. In geval van dissectie of ruptuur is uiteraard wel een dringende ingreep noodzakelijk, doch dit gaat met een duidelijk hoger risico gepaard.
De prognose van Marfan syndroom is wisselend en sterk afhankelijk van de aard en de ernst van de hartaantasting. Bij vrouwen, zelfs met een milde cardiale aantasting, is de zwangerschap een erg risicovolle periode. Een mogelijke zwangerschap dient dan ook goed gepland en gevolgd worden, zowel door de cardioloog als de gynaecoloog.
'Geef Hartekind een gezicht' en deel je verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
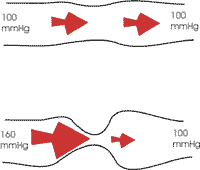
Pulmonalisstenose behoort tot de frequent voorkomende aangeboren hartafwijkingen. Het is een verdikking of vergroeiing van de klep tussen de rechterkamer en longslagader, waardoor deze onvoldoende opent om het bloed naar de longen te doen stromen. Hierdoor moet de rechterkamer meer druk uitoefenen om het bloed toch door die vernauwde klepopening te stuwen. De continu verhoogde druk veroorzaakt een verdikking van de hartspier (vergelijkbaar met atleten die voortdurend grote inspanningen leveren). Uiteindelijk geeft dit op lange termijn ook vermoeidheid en aftakeling van de hartspier.
Meestal hebben kinderen met deze afwijking nauwelijks klachten. Er is bijna altijd een hartgeruis dat uiteindelijk tot verder onderzoek zal leiden. Alleen pasgeborenen met een ernstige verdikking van deze klep vertonen symptomen.
De diagnose wordt verkregen door een echocardiografie. Deze brengt de verdikte klep in beeld. Doppler-technieken laten ons toe het drukverschil over de klep te meten.
Een behandeling wordt ingesteld vanaf een drukverschil van 45 mm Hg of indien er klachten zijn die toegeschreven kunnen worden aan de klepverdikking. Men weet uit ervaring dat indien men in die omstandigheden niet ingrijpt, het risico op hartspierbeschadiging bestaat. De behandeling bestaat uit een hartkatheterisatie met ballondilatatie. Deze wordt meestal uitgevoerd onder algehele anesthesie.
Het risico van de ingreep is zeer laag en er zijn weinig complicaties. Alleen bij pasgeborenen is de ingreep iets delicater. De resultaten zijn meestal zeer goed met volledige genezing bij meer dan 90% van de behandelde patiënten. Slechts uitzonderlijk is een tweede dilatatie nodig. In een klein aantal gevallen is de klep echter vergroeid en verdikt, maar eveneens zeer elastisch (dysplastisch). Meestal komt dat voor bij kinderen die ook andere afwijkingen vertonen zoals syndroom van Noonan. In die gevallen slaagt men er met ballondilatatie niet in de vergroeiing open te blazen. Enkel openhart chirurgie kan dan een oplossing bieden. Gelukkig zijn de risico’s van dit type operatie laag en de resultaten goed.
Na een geslaagde en tijdige ingreep verwacht men voor deze kinderen een normaal leven, sportbeoefening incluis.
Indien er kinderwens is, is het belangrijk om de pulmonalisstenose te behandelen vooraleer een zwangerschap te laten aanvangen.
'Geef Hartekind een gezicht' en deel je verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Bij pulmonale hypertensie of “hoge bloeddruk in de longen” is de weerstand in de longbloedvaten verhoogd. In het begin van de ziekte wordt dit veroorzaakt door het overdreven samentrekken van de bloedvatwand waardoor de doormeter van het bloedvat verkleint. In dit vroege stadium kan men de bloedvaten nog doen ontspannen door het toedienen van medicatie. Als de situatie echter lang blijft bestaan, gaat de bloedvatwand verdikken (hypertrofie) waardoor de binnendoormeter van het bloedvat definitief nauwer wordt en de weerstand die het hart moet overwinnen om het bloed door die vernauwde bloedvaten te kunnen pompen verhoogt. Eens zover, kan de toegenomen weerstand niet meer met medicatie genormaliseerd worden. Door de verhoogde longvaatweerstand moet de rechterkamer dus een grotere druk genereren om het bloed naar de longen te pompen. De spierwand van de rechterkamer verdikt en uiteindelijk gaat de rechterkamer ook uitzetten (dilateren). Dit leidt uiteindelijk tot rechterhartfalen.
Pulmonale hypertensie heeft verscheidene oorzaken. Bij kinderen zijn er 3 belangrijke:
Men weet dat bij kinderen verschillende oorzaken van pulmonale hypertensie samen kunnen voorkomen, wat de behandeling soms zeer ingewikkeld maakt. Ook bepaalde genetische afwijkingen zoals Downsyndroom verhogen het risico op pulmonale hypertensie op kinderleeftijd.
Patiënten met pulmonale hypertensie hebben vooral klachten van vermoeidheid, kortademigheid en een verminderd inspanningsvermogen. Als er een verbinding bestaat tussen het linker- en het rechterhart zal, door de verhoogde drukken rechts, bloed van het rechter (zuurstofarme) naar het linker (zuurstofrijke) hart stromen (Eisenmenger complex). Deze patiënten krijgen een blauwe verkleuring van huid en lippen (cyanose).
Als de diagnose van pulmonale hypertensie wordt verdacht, is een volledige uitwerking nodig om de ernst, oorzaak en prognose te bepalen. Echografie van het hart kan de rechterkamerafwijkingen aantonen en de longdruk kan worden ingeschat. Hartkatheterisatie laat toe de druk in de rechterkamer en de longvaatweerstand te meten. Tijdens de hartcatheterisatie worden soms stoffen die de longvaten doen ontspannen toegediend. De reactie hierop laat ook toe om te bepalen of de pulmonale hypertensie nog omkeerbaar is of niet. Afhankelijk van het klinisch beeld worden een CT scan van de longen, ventilatie-perfusiescan, stollingsonderzoek, levertesten…. uitgevoerd om andere oorzaken van pulmonale hypertensie op te sporen.
De behandeling van pulmonale hypertensie is voornamelijk medicamenteus. Acute opstoten van pulmonale hypertensie waarbij opname en beademing van de patiënt nodig zijn, worden behandeld met inhalatie via het beademingstoestel van stikstofmonoxide (NO). Chronische behandeling bestaat voornamelijk uit calciumblokkers bij diegenen die goed reageren op de stoffen die tijdens de hartcatheterisatie worden toegediend (reversiebele vorm). Ondertussen zijn er ook andere medicamenten van dezelfde families beschikbaar. Als er onvoldoende respons is op medicatie in pilvorm kan overgeschakeld worden op medicatie die langs een ader wordt toegediend via een continu pompsysteem (prostaglandines). Indien dit onvoldoende is blijft een hart- en longtransplantatie de enige optie.
De prognose van deze pulmonale hypertensie is afhankelijk van het stadium en de oorzaak. Als er een onderliggende cardiale afwijking bestaat die chirurgisch kan gecorrigeerd worden, is, mits de operatie tijdig wordt uitgevoerd, de pulmonale hypertensie omkeerbaar. Is er echter al een evolutie naar Eisenmengercomplex, is dit niet meer mogelijk. Bij primaire pulmonale hypertensie is het proces steeds progressief maar kan de evolutie wel vertraagd worden door de beschikbare medicatie.
Bij vrouwelijke patiënten is de aanwezigheid van pulmonale hypertensie een ABSOLUTE contra-indicatie voor zwangerschap. Voor de geschikte anticonceptie moet gezamenlijk advies van cardioloog/pneumoloog en gynecoloog ingewonnen worden omdat sommige vormen van anticonceptie gevaarlijk kunnen zijn.
'Geef Hartekind een gezicht' en deel je verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Pulmonalisatresie met intact interventriculair septum is een ernstige aangeboren hartafwijking waarbij de longslagaderklep abnormaal is aangelegd of volledig ontbreekt. Hierdoor kan het bloed vanuit de rechterkamer niet naar de longen stromen. Bovendien zijn er meestal ook afwijkingen ter hoogte van de tricuspidalisklep en is de rechterkamer te klein (hypoplastisch).
Afwijkingen ter hoogte van de kransslagaders zijn frequent aanwezig en bepalen in hoge mate de prognose en de behandelingsmogelijkheden.
Door de afwezigheid van een normale longslagaderklep kan het zuurstofarme bloed niet vanuit het hart naar de longen stromen. Dit leidt tot ernstige blauwheid (cyanose) tijdens de eerste levensdagen.
'Geef Hartekind een gezicht' en deel je verhaal.
Tetralogie van Fallot bestaat uit vier “componenten”: (1) een groot gat tussen de twee kamers; (2) de aorta “te paard” over het defect; (3) een te smalle uitgang van de rechter kamer door vernauwingen onder (infundibulaire stenose), op (valvulaire stenose) of boven de klep (supravalvulaire stenose) of in de longtakken (takstenose); (4) een verdikte spierwand van de rechter kamer (hypertrofie).
Er zijn heel veel varianten. De klachten zijn afhankelijk van de graad en plaats van de vernauwing naar de longen toe. Hoe nauwer de uitgang naar de longen, hoe vroeger en hoe ernstiger blauwheid zal worden opgemerkt. Als de uitgang naar de longen helemaal afwezig is, spreekt men van “Fallot met pulmonale atresie”. Indien er weinig obstructie is, zal de blauwheid niet uitgesproken zijn en spreekt men van “Witte” Fallot. Bevindt de obstructie zich vooral ter hoogte van de spier onder de longklep, dan bestaat het gevaar op plotse aanvallen van blauwheid door kramp van deze spierring: er gaat bijna geen bloed meer naar de longen waardoor de baby wit-blauw wordt, gaat zweten en vaak bewusteloos wordt. Dit zijn de “Fallot-spells”. Tijdstip van aanval of uitlokkende factoren zijn verscheiden. Wat ouders zelf kunnen doen is het kind troosten en de knieën van het kind tegen de borst duwen om zoveel mogelijk bloed naar de longen te stuwen. Meestal gaat de aanval daarmee over. Uw kindercardioloog moet zeker van dit type aanvallen op de hoogte gebracht worden. Eén cyanotische aanval is reeds voldoende reden om tot een chirurgische ingreep over te gaan.
Het tijdstip van de behandeling hangt af van de ernst van de blauwheid. Onder de leeftijd van 3 maanden wordt geopteerd voor een tijdelijke oplossing zoals de Blalock-Taussig shunt (aanleggen van verbindingsvat tussen lichaamsslagader en longsslagader) of ballondilatatie van de pulmonaalklep. Boven de leeftijd van 3 maanden wordt meestal gekozen voor een totale correctie. Indien er geen klachten zijn wordt deze correctie uitgevoerd tussen 6 en 12 maanden. Het risico van de ingreep is laag maar restletsels zijn niet zeldzaam. Meestal gaat het om een lek van de pulmonaalklep of vernauwingen van de longslagaders. Toch is de levenskwaliteit van de meeste kinderen zeer goed: zij kunnen normaal naar school en deelnemen aan sportactiviteiten. Op volwassen leeftijd moeten sommigen omwille van een lekkende pulmonaalklep een heringreep ondergaan met implantatie van een nieuwe pulmonaalklep. Dit kan zowel chirurgisch als interventioneel.
Indien er ernstige vernauwingen bestaan in de longslagaders kunnen deze opgerokken worden met een ballondilatatie of stentimplantatie.
Sem, Lyvv, Jimi, Denise, Noa, Hessel en Gio hebben Tetralogie van Fallot, lees hun verhaal bij 'Geef Hartekind een gezicht'
Indien de longslagadertakken normaal zijn aangelegd, gebeurt de bloedvoorziening naar de longen meestal via de ductus arteriosus (verbindingsvat tussen aorta en longslagader). Vaak echter bestaat er geen hoofdstam van de longslagader en de bloedvoorziening naar de longen gebeurt dan via de zogenaamde MAPCAs, abnormale bloedvaten die meestal ontspringen op de aorta en afzonderlijk de verschillende delen van de long bevloeien.
'Geef Hartekind een gezicht' en deel je verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Bij totaal abnormale pulmonaalveneuze uitmonding monden de longvenen, via een verbindingvat (“collector vene”), uit in de rechter- in plaats van in de linkervoorkamer. Meestal bestaat er ook een opening tussen beide voorkamers waardoor het zuurstofarme en het zuurstofrijke bloed zich met elkaar kunnen vermengen. Er worden verschillende soorten TAPVU beschreven, afhankelijk van de plaats waar de longvenen in de rechtervoorkamer toekomen. Men spreekt van een supracardiale vorm (via vena cava superior), infradiafragmatische vorm (via vena cava inferior) en intracardiale vorm (rechtstreeks in hart). Door deze abnormale uitmonding gaat de rechtervoorkamer uitzetten en de rechterkamer dilateert. Uiteindelijk worden ook de longen overbelast.
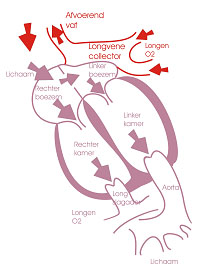
Vaak treden er slechts klachten op na enkele weken. De voeding verloopt moeizaam en de gewichtsevolutie is meestal onbevredigend. De ademhaling is versneld (tachypneu). Indien er een vernauwing bestaat op de longvenen of ter hoogte van de uitmonding treden er wel symptomen op onmiddellijk na de geboorte. De baby vertoont ademhalingsproblemen en heeft een blauwe kleur. Snel chirurgisch ingrijpen is dan noodzakelijk.
De behandeling van abnormale longvene uitmonding is chirurgisch. De collectorvene wordt tegen het linker atrium gebracht en er wordt een nieuwe verbinding (“anastomose”) tussen beiden aangelegd. De collectorvene wordt afgebonden.
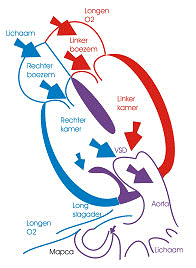
Transpositie van de grote vaten is een hartgebrek waarbij de aansluiting van aorta en longslagader verkeerd is. In een normale situatie staat de aorta boven de linker kamer en de longslagader boven de rechter kamer. Bij transpositie is dit net andersom.
![]()
Er wordt een onderscheid gemaakt tussen de “klassieke” transpositie, waarbij er geen andere belangrijke afwijkingen aanwezig zijn, en de “transpositiecomplexen”, waarbij er naast de abnormale aansluiting van de grote slagaders ook andere afwijkingen aan het hart voorkomen (een groot gat tussen de kamers, vernauwde of volledig afgesloten kleppen, enz…)
In de klassieke transpositie met intact septum bestaat er een parallelle bloedcirculatie: het zuurstofrijke (“rode”) bloed dat uit de longen komt vloeit terug naar de longen en het zuurstofarme (“blauwe”) bloed uit het lichaam terug naar het lichaam; Indien er geen verbindingen bestaan tussen longdoorstroming en lichaamsdoorstroming, is deze situatie niet leefbaar omdat het lichaam zo geen zuurstofaanvoer meer krijgt. Gelukkig zijn er bij de geboorte meestal wel verbindingen aanwezig: zowel ter hoogte van het tussenschot tussen de voorkamers (het “ovale venster”) als ter hoogte van de zogenaamde ductus van Botalli, een kort verbindingsvat tussen de aorta en de longslagader. Door het bestaan van deze verbindingen wordt de blauwheid van het kind meestal pas enkele uren na de geboorte zichtbaar. Op dit ogenblik zijn we in staat deze bestaande verbindingen open te houden tot het ogenblik van de chirurgische totale correctie. De ductus wordt opengehouden door het geven van medicatie. Het ovale venster wordt via hartkatheterisatie opengescheurd (ballonprocedure volgens Rashkind).
Klassieke transpositie wordt nu zo snel mogelijk volledig gecorrigeerd. De chirurgische correctie wordt uitgevoerd met een laag risico. Vooral abnormaal verlopende kransslagaders kunnen het operatief risico beïnvloeden. Als de operatie geslaagd is, zijn levensverwachting en -levenskwaliteit normaal.
Na de chirurgische correctie moeten de kinderen verder opgevolgd worden met vooral aandacht voor eventuele vernauwingen op de longslagader. Ernstige kleplekken of kransslagadervernauwingen zijn zeldzaam.
Vroeger ondergingen kinderen met transpositie een ander type operatie waarbij niet de slagaders maar de boezems werden “verwisseld”: Senning of Mustard operatie. Kinderen met dit type ingreep hebben later een groter risico op ritmestoornissen en laattijdig hartfalen.
Transpositie is één van de hartgebreken die makkelijk gemist kunnen worden bij de foetale screening
Lees de verhalen van James, Berber, Rijk en Jord. Zij hebben transpositie van de grote vaten.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
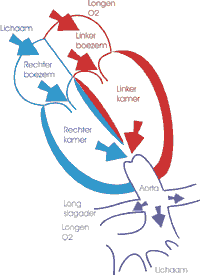
Lees de verhalen van Michella en van Julia, geboren met truncus arteriosus.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
Zelden zijn hartafwijkingen de oorzaak van aanslepende ademhalingsproblemen bij oudere kinderen, tenzij ze druk veroorzaken op de luchtwegen. Hierdoor ontstaat een wisselende vernauwing van de luchtpijp, waardoor ademen moeilijker wordt. Vooral bij inspanning of infectie zullen de problemen verergeren. Hartafwijkingen die dit veroorzaken worden samengebracht onder de noemer “vasculaire ring”.
Normaal liggen aortaboog en longslagader vrij in de borstkas en kruisen de luchtwegen slechts aan één zijde. Soms is de aanleg van deze bloedvaten gestoord en lopen zij voor en achter de luchtpijp. Ze vormen zo een ringstructuur die de luchtpijp en vaak ook de slokdarm vernauwt. Afhankelijk van de strakheid van de ringstructuur ontstaan ademhalingsproblemen op zuigelingen- of op kinderleeftijd.
De behandeling bestaat steeds uit chirurgie. Bij aortaboogafwijkingen volstaat een vrij eenvoudige operatie die meestal via een sneetje in de zijkant van de linker borsthelft uitgevoerd wordt en waarbij men het ligament of de overtollige linker aortaboog doorsnijdt. Dit is geen open hartoperatie en het operatief risico is zeer laag. Voor de abnormale linkerlongslagader moet een klassieke open hartoperatie met openen van het borstbeen uitgevoerd worden. Het risico van de operatie is laag.
Na behandeling kan men bij grotere kinderen een snelle verbetering van de klachten verwachten. Sommigen blijven wel gevoelige luchtwegen behouden, maar deze kunnen gemakkelijker geholpen worden met klassieke medicatie. Bij jongere kinderen kunnen de problemen nog gedurende een zestal maanden aanslepen. Nadien zijn ze dan meestal definitief genezen. Alleen bij een deel van de zuigelingen met een abnormaal verlopende longslagader kan een zodanige misvorming van de luchtpijp bestaan dat zij na de operatie niet meer zelfstandig kunnen ademen. Bij deze kinderen is er een verhoogd risico op overlijden ondanks een geslaagde operatie.
'Geef Hartekind een gezicht' en deel je verhaal.
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blanquaert.
Het geïsoleerde VSD is de meest voorkomende aangeboren hartafwijking. Er bestaan eén of meerdere openingen in het tussenschot tussen beide kamers waarlangs het bloed van de linker naar de rechter kamer stroomt.
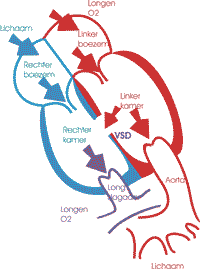
Aanvankelijk is die bloedstroom beperkt omdat voor de geboorte de druk in rechter en linkerkamer gelijk is. De longen zijn nog niet ontplooid en via de ductus arteriosus staat de longcirculatie (en dus ook de rechterkamer) in verbinding met de systeemcirculatie. In beide systemen heerst dezelfde druk en er is geen passage van bloed door het VSD. Na de geboorte ontplooien de longen en sluit de ductus. Dit heeft tot gevolg dat in de eerste dagen na de geboorte de longweerstand en de rechter kamerdruk dalen. Er ontstaat een drukverschil tussen linker en rechter kamer waardoor het bloed onder hoge druk doorheen het VSD wordt geperst. De turbulentie die ontstaat zorgt voor een hartgeruis, dat typisch bij ontslag uit het moederhuis gehoord wordt.
Klachten treden op naargelang de grootte van het defect. Een klein defect zal gekenmerkt worden door een luid geruis maar geen klachten geven. Bij een groot defect (meestal ³1 cm) zal er veel bloed vanuit de linker naar de rechter kamer en van daaruit naar de longen stromen. Vanaf de geboorte zakt de longweerstand geleidelijk tot zijn minimum rond de 2e tot de 3e levensmaand. Op dat ogenblik zal de bloedstroom doorheen het VSD naar de longen toe maximaal worden en treden er klachten op: snelle ademhaling tijdens het drinken en overvloedig zweten na het drinken. Geleidelijk aan zullen ook de hoeveelheden die het kind drinkt verminderen en de gewichtstoename vertraagt. Algemeen ziet het kind bleek of grauw. Cyanose is geen kenmerk van deze afwijking. Wel is er meestal een luid hartgeruis.
Kleine defecten worden rustig gevolgd. De kans op spontane sluiting voor het 6e levensjaar is zeer groot (meer dan 90%). Indien de opening na deze leeftijd nog steeds aanwezig is, wordt enkel antibiotica profylaxe aangeraden. Operatief ingrijpen bij een klein VSD is enkel nodig indien een klep in de onmiddellijke nabijheid lekt.
Grote defecten die klachten geven, kunnen eerst met medicatie behandeld worden (diuretica) in de hoop dat ze spontaan verkleinen.
Indien de klachten aanslepen, dient het VSD chirurgisch te worden gesloten. Deze ingreep gebeurt klassiek tijdens het eerste levensjaar. Indien de klachten echter verbeteren, kan men enige tijd afwachten. Tijdens de daarop volgende periode worden voornamelijk de longdruk en de grootte van het hart gevolgd. Indien de longdruk verhoogt, zal men, zelfs zonder klachten, het VSD chirurgisch sluiten voor de eerste verjaardag en dit om onherstelbare schade aan de longvaten te voorkomen. Indien de longdruk normaal blijft, maar het hart is rond de leeftijd van 5 jaar vergroot, zal men het VSD rond deze leeftijd sluiten. Het operatief risico van deze ingreep is laag en de levensverwachting nadien is normaal.
Een alternatief wijze van sluiting met een paraplusysteem via hartkatheterisatie is in uitzonderlijke gevallen mogelijk: slechts openingen in het midden van het tussenschot en ver verwijderd van de kleppen kunnen met deze techniek gesloten worden.
Soms zijn er verscheidene VSD’s in de punt van het hart. In tegenstelling tot VSD’s op andere locaties is chirurgische sluiting hier niet zo eenvoudig. Dikwijls kan men deze openingen slechts benaderen via een insnede in het linkerhart, met nadien een zeker risico op blijvende beschadiging van het linkerhart. De voorkeursbehandeling bestaat nu uit sluiten van deze openingen met catheterisatietechnieken, zoals bij ASD. Soms legt men tijdelijk een bandje rond de longslagader, om wat tijd te winnen. In sommige gevallen zullen de VSD’s hieronder zelfs spontaan sluiten. Daarom worden nu zelfs resorbeerbare bandjes ontwikkeld.
Lees hier het verhaal van Nina
Bron: Centrum voor Aangeboren Hartafwijkingen Anna Blancquaert
De ziekte van Ebstein is een zeer zeldzame aangeboren hartafwijking. De tricuspiedklep (tussen rechterboezem en rechterkamer) is naar de punt van de rechterkamer verplaatst en hangt gedeeltelijk vast aan het tussenschot. De rechterkamer wordt daardoor kleiner en pompt het bloed minder efficiënt naar de longen.
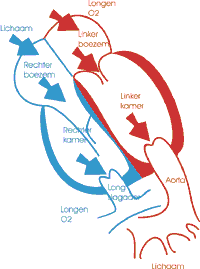
'Geef Hartekind een gezicht' en deel je verhaal.
De ziekte van Kawasaki is een zeldzame systeemziekte. Ongeveer 50% van de gevallen treedt op voor de leeftijd van 2 jaar. De juiste oorzaak is nog steeds niet gekend, doch de ziekte berust waarschijnlijk op een reactie van het immuniteitssysteem op een uitwendige factor (infectie, giftig product,…). Ze is niet besmettelijk en voor zover men weet ook niet erfelijk.
Kinderen met de ziekte van Kawasaki zijn erg ziek. Ze hebben hoge koorts die meer 5 dagen duurt, rode en jeukende ogen, rode mondslijmvliezen en vaak ook gebarsten lippen, rode huiduitslag die afschilfert ter hoogte van tenen en vingers en gezwollen lymfeklieren. Bij kinderen jonger dan 1 jaar zijn soms slechts enkele van deze tekens aanwezig. Naast de uitwendig zichtbare afwijkingen, kan er ook een ontsteking optreden ter hoogte van het hart, meer bepaald ter hoogte van de kransslagaders.(coronaire arteriën) Deze kunnen hierdoor locaal uitzetten en aneurysmata vormen. In deze aneurysmata kunnen er zich vervolgens klonters (thrombi) vormen die de bloedvoorziening van het hart bedreigen en aanleiding kunnen geven tot een hartinfarct.